
Earlier CAGE-based projects consisted of sequencing 50-100,000 tags/library and this allowed mapping of the promoter of the main transcripts. However, it quickly became clear that there are many more TSSs than the number of genes (>230,000 TSSs in the mouse. Additionally, for statistically relevant expression measurement for specific promoters, it is necessary to sequence a given RNA end multiple times in order to count and statistically evaluate the promoter usage. For this, we have adapted CAGE to 2nd generation sequencing as it has become available in Japan. We have adapted CAGE to the 454 Life Science sequencer[1] to create what we have coined deepCAGE[2] [3]. DeepCAGE technology does not require cloning the tag concatamers in plasmids.
DeepCAGE is based on priming total RNA in the first strand cDNA reaction with random primers to capture both the polyA+ and polyA- RNAs species. Oligo-dT priming is an option for particular usages, but is not generally recommended for expression analysis. To extend cDNA synthesis through GC-rich regions in the 5’ UTR, we carry out reverse transcription reactions at high temperature in the presence of trehalose and sorbitol8. cDNAs reaching the cap-site are selected by cap-trapping and ligated to a linker having a recognition site for the class-IIs restriction endonuclease MmeI (or more recently, EcoP15I, which cleaves 27 nt apart), right next to the start of the cDNAs corresponding to the 5’ end of the original RNAs. This linker is used to prime second-strand cDNA synthesis. Subsequently, MmeI (EcoP15I) digestion cleaves 20~21 (or 27) bp within the double-stranded cDNA, releasing CAGE tags. After ligation of a second linker to the 3’ end opened by MmeI or EcoP15I digestion, CAGE tags are PCR-amplified. In earlier procedures based on sequencing using the 454 Life Sciences sequencer, we used concatenation. Typically, a run of the 454 produces 750,000 to a million CAGE tags; such a large amount of 454-deepCAGE data was produced in the Genome Network/FANTOM4 project.
DeepCAGE with Solexa and SOLiD
To further decrease the cost of sequencing, we adapted deepCAGE to the Illumina (Solexa) sequencing technology and the ABI SOLiD sequencers that have appeared more recently on the market. The technical changes are minor compared to the first version of deepCAGE, when the primer sequences had to be adapted for different sequencing primer designs on the Illumina GA/GAII and SOLiD and tedious concatamer formation was eliminated. The advantages of these sequencers over the 454 are as follows: (a) production of concatamers is unnecessary, so there are fewer PCR cycles: 13-15 PCR cycles compared to 25 or more. This reduces PCR bias; (b) decreased cost/tags, because 1 run with the Solexa GA II produces more than 50 millions tags, and the SOLiD produces >100 millions per run; (c) in this development we have also switched from CAGE tags that are 20 nt long to tags that are 27 nt long, increasing the mapping rate from 65-70% to 80-85%, and increasing the chance of detecting RNAs originating in gene families and genomic regions that are very similar.
Tags of short stretches of DNA are preferable to longer molecules for promoter usage profiling because short tags of 27 nt inserts, like in CAGE, do not to show size bias during the PCR and heat denaturation and primer extension are less likely to affect them. On the contrary, the amplification of long, heterogeneously sized full-length cDNAs does surely show size bias. For instance the amplification of 5’ ends of full-length cDNAs (or even randomly primed 500-1000 bb 5’ cDNAs ends) severely biases the quantitative representation of original transcripts based on length and GC content. Additionally, Solexa sequencing requires the bridge PCR step of the DNA molecules attached to the sequencing slides. Bridge PCR is more efficient for short inserts than for very long inserts (with the upper limitation of about 1 Kb): this excludes most of the full-length cDNAs. The analog amplification step for SOLiD and 454 is the emulsion PCR, which is limited to fragments of similar size (~1Kb). Technologies for sequencing from both ends are important for mapping the transcriptome borders, but show bias that is not recommended when the intent is to detect transcriptional activity at a given promoter.
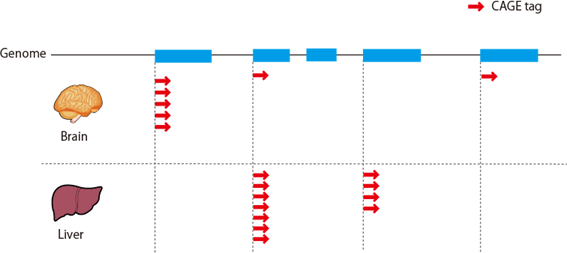
Figure 1: DeepCAGE defines transcription starting site and it frequency
References
- ^ Maeda, N. et al. Development of a DNA barcode tagging method for monitoring dynamic changes in gene expression by using an ultra high-throughput sequencer. Biotechniques 45, 95-7 (2008), doi: 10.2144/000112814
- ^ de Hoon M, Hayashizaki Y Deep cap analysis gene expression (CAGE): genome-wide identification of promoters, quantification of their expression, and network inference. Biotechniques 2008 Apr;44(5):627-8, 630, 632, doi: 10.2144/000112802
- ^ Valen et al. Genome-wide detection and analysis of hippocampus core promoters using DeepCAGE Genome Research 19:255-265 2009, doi: 10.1101/gr.084541.108