
An atlas of human long non-coding RNAs with accurate 5’ ends
Chung-Chau Hon, Jordan A. Ramilowski, Jayson Harshbarger, Nicolas Bertin, Owen J. L. Rackham, Julian Gough, Elena Denisenko, Sebastian Schmeier, Thomas M. Poulsen, Jessica Severin, Marina Lizio, Hideya Kawaji, Takeya Kasukawa, Masayoshi Itoh, A. Maxwell Burroughs, Shohei Noma, Sarah Djebali, Tanvir Alam, Yulia A. Medvedeva, Alison C. Testa, Leonard Lipovich, Chi-Wai Yip, Imad Abugessaisa, Mickaël Mendez, Akira Hasegawa, Dave Tang, Timo Lassmann, Peter Heutink, Magda Babina, Christine A. Wells, Soichi Kojima, Yukio Nakamura, Harukazu Suzuki, Carsten O. Daub, Michiel J. L. de Hoon, Erik Arner, Yoshihide Hayashizaki, Piero Carninci & Alistair R. R. Forrest
Nature 2017
DOI: 10.1038/nature21374
Long non-coding RNAs (lncRNAs) are largely heterogeneous and functionally uncharacterized. Here, using FANTOM5 cap analysis of gene expression (CAGE) data, we integrate multiple transcript collections to generate a comprehensive atlas of 27,919 human lncRNA genes with high-confidence 5′ ends and expression profiles across 1,829 samples from the major human primary cell types and tissues. Genomic and epigenomic classifications of these lncRNAs reveals that most intergenic lncRNAs originate from enhancers rather than from promoters. Incorporating genetic and expression data, we show that lncRNAs overlapping trait-associated single nucleotide polymorphisms are specifically expressed in cell types relevant to the traits, implicating these lncRNAs in multiple diseases. We further demonstrate that lncRNAs overlapping expression quantitative trait loci (eQTL)-associated single nucleotide polymorphisms of messenger RNAs are co-expressed with the corresponding messenger RNAs, suggesting their potential roles in transcriptional regulation. Combining these findings with conservation data, we identify 19,175 potentially functional lncRNAs in the human genome.

Figure 1. We used a technology known as Cap Analysis of Gene Expression (CAGE), which was developed at RIKEN, to build an atlas of human long non-coding RNAs with accurate 5’ ends, precisely pinpointing where in the genome their transcription is initiated. We then integrated the FANTOM5 CAGE datasets with transcript models from diverse sources, and built a meta-assembly called FANTOM CAGE associated transcriptome (FANTOM CAT). Comparing with GENCODE, FANTOM CAT contains 19,688 more lncRNA loci and their 5’ ends are better supported with epigenome evidence.
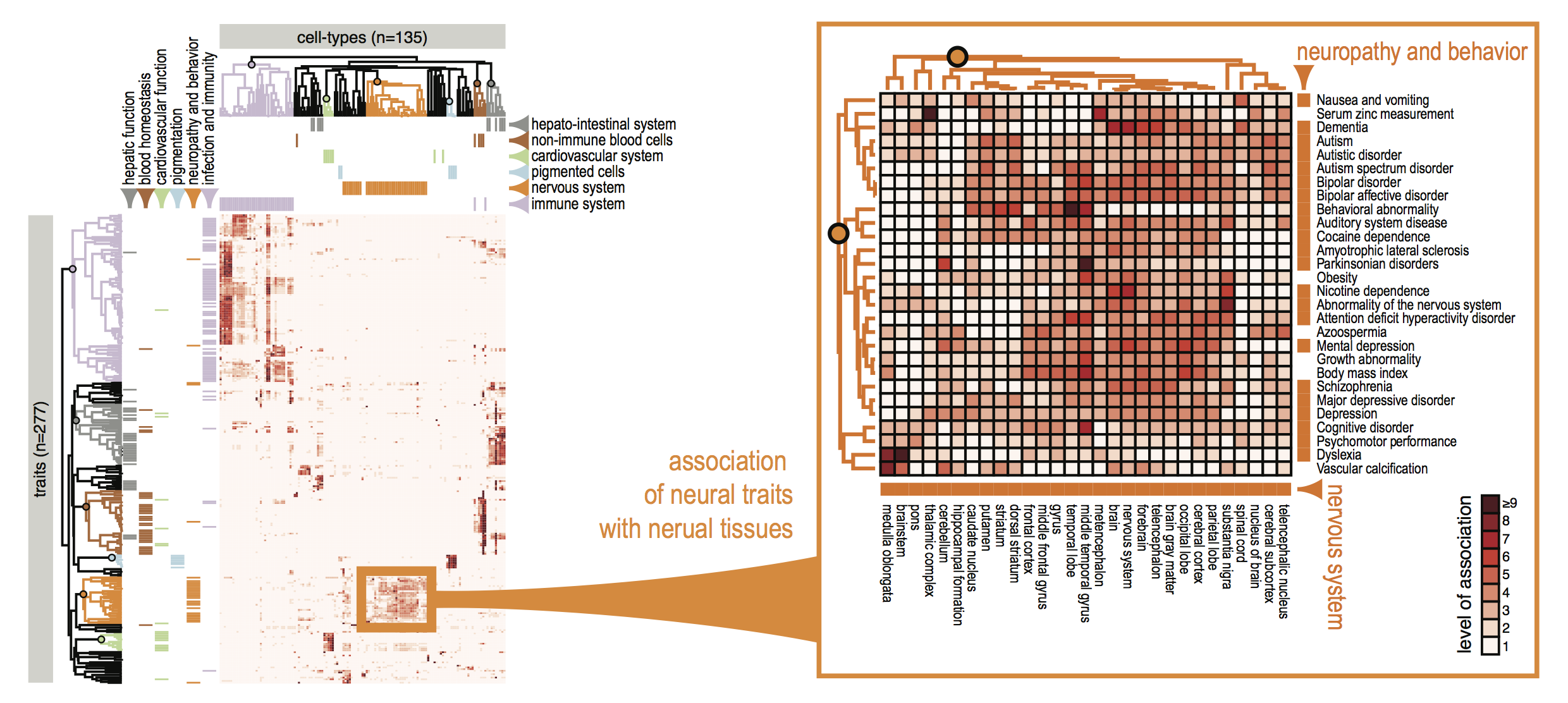
Figure 2. FANTOM CAT genes were 1) associated with traits based on GWAS SNP, and 2) associated with cell-types based on sample ontology enrichment analysis. On the basis of these lists of cell-type-enriched and trait-associated genes, we evaluated the association between 345 cell types and 603 traits (208,035 possible pairs) and identified 1,874 pairs of cell types and traits with significant association. Unsupervised clustering of significantly associated cell-type–trait pairs revealed that related cell types and traits tended to cluster together.
FANTOM CAT Browser
- FANTOM CAT Browser - an web application allows users to browse genes, view their genomic loci through ZENBU, filter by their annotations, intersect them with their associated sample ontologies or traits, and download the relevant data
Zenbu Views
- Minimal - displays minimal information, including CAGE signal, gene, transcripts and conservation
- Main - displays essential information, e.g. expression, conservation and SNPs and more
- Expression - focus on expression profile, e.g. expression levels, sample ontology enrichment, dynamic expression and more
- Assembly - focus on assembly information, e.g. source transcripts, FANTOM CAT at other cutoffs and more
- Epigenome - focus on epigenomic information, e.g. Roadmap chromHMM state and histone marks
- Evolution View - focus on evolutionary information, e.g. RS score, orthlogous expression in other species
- Extended - displays integrated information and users can turn on extra tracks for elaborated information
Data
Direct link to this directory:
| Name | Filesize | Last Modified |
|---|---|---|
| 5.04 kb | Aug 09, 2017 | |
| Files: 1 | Directories: 5 | ||
Questions
Having trouble with FANTOM CAT? Please ask us on fantom-cat-users Google user group or send us an email.